
Introduction
RNA-Seq variant calling provides a powerful approach for uncovering genetic variations, understanding gene regulation, and identifying potential disease-associated variants. Its ability to capture transcriptome-wide information makes it a valuable tool for basic research and clinical applications. RNA-Seq variant calling offers several advantages, making it a useful tool for genomic research and clinical applications. SNP detection by RNA-seq is particularly interesting for various species since whole genome sequencing is expensive and exome sequencing tools are unavailable. In addition, applying deep-learning techniques to RNA-seq variant calling has dramatically increased the chances of identifying variants from RNA-seq data [1].
Zifo Bioinformatics team has experience in working on RNA-seq variant calling, which has applications in diverse fields such as:
RNA-Seq Variant calling - Challenges and considerations:
Some of the key challenges and considerations in RNA-seq pipeline building are given below, all of which the Zifo bioinformatics team has handled.
Addressing these challenges requires specialized bioinformatics tools, careful quality control measures, validation using independent methods, and consideration of the specific characteristics of RNA-Seq data.
Based on Zifo's experience and knowledge derived from working on multiple projects we see the cost of RNA-Seq can range from a few hundred dollars to several thousand dollars per sample. As technology advances and economies of scale improve, the price of RNA-Seq has decreased over time. It is crucial to consult with sequencing service providers or core facilities to obtain accurate cost estimates tailored to your specific project requirements. They can provide detailed information on sequencing platforms, library preparation options, and associated costs.
In addition, the Zifo BIO team can help in RNA-seq pipeline development (Figure 1.) related to Preprocessing (quality control, reads alignment), post-processing, variant calling (selecting RNA-Seq variant callers and parameter customization) and reporting. Furthermore, Metrics Capture during pipeline execution is an additional service performed by the Zifo BIO team. It provides valuable data to the scientific team and the IT department, and the infrastructure teams.
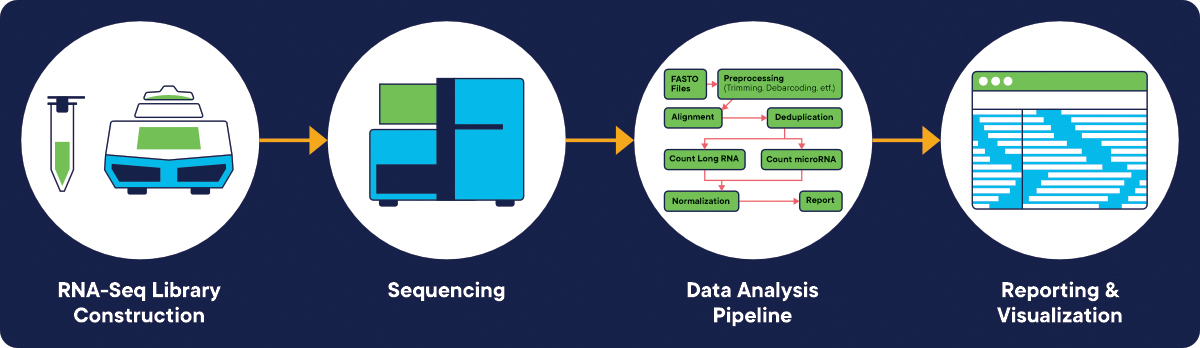
Figure 1. RNA-seq pipeline workflow starting from library construction, sequencing, Bioinformatics pipeline (development and execution) finally ending with reporting, data analysis and results visualization. [Image credit: www.bio-rad.com].
Zifo's experience in the field of RNA-Seq Variant calling in Cancer and Normal samples
RNA-Seq variant calling in cancer and normal samples can provide valuable insights into somatic mutations specific to cancer cells. Here are some considerations when performing variant calling with cancer and normal RNA-Seq samples:
Since the normal sample contains germline variants in the patient's genome, it is important to filter out these variants from the variant calling results. Tools like GATK's "MuTect" or "VarScan" offer specific options for identifying somatic variants by filtering out germline variants in the normal sample..
It's important to note that RNA-Seq variant calling in cancer samples may have additional complexities due to tumour heterogeneity, sub-clonal mutations, and variations in gene expression levels. These challenges may require specialized methods for accurate variant calling and interpretation. Overall, performing variant calling with cancer and normal RNA-Seq samples can provide insights into the somatic mutations specific to cancer cells, enabling a better understanding of the genetic alterations underlying cancer development and progression.
If you're interested in RNA-Seq variant calling design, custom enhancement, research, and development, contact us by email at info@zifornd.com, and we are eager to collaborate and discuss further.
Reference: [1]- (A deep-learning-based RNA-seq germline variant caller) https://academic.oup.com/bioinformaticsadvances/article/3/1/vbad062/7197031?login=false#409987722